Tabla 1. Clasificación clínica actualizada de hipertensión pulmonar 2018

ARTICULO DE ACTUALIZACION
La necesidad de la detección temprana
de la hipertensión arterial pulmonar
Raúl J. Bevacqua1, Sergio V. Perrone2
1 Médico cardiólogo. División Cardiología. Pabellón Inchauspe. Hospital General de Agudos “Dr. J. M. Ramos Mejía”. GCBA.
Ciudad Autónoma de Buenos Aires. República Argentina.
2 Médico cardiólogo. Instituto FLENI. Ciudad Autónoma de Buenos Aires. República Argentina.
Instituto Argentino de Diagnóstico y Tratamiento. Ciudad Autónoma de Buenos Aires. República Argentina.
Hospital de Alta Complejidad en Red “EL Cruce” Néstor Kirchner. Florencio Varela. Buenos Aires. República Argentina.
Universidad Católica Argentina. Ciudad Autónoma de Buenos Aires. República Argentina.
Correspondencia: Dr. Raúl J. Bevacqua.
E-mail: raulbev@hotmail.com
Recibido: 28/12/2021
Aceptado: 10/02/2022
Resumen
La hipertensión arterial pulmonar (HAP), Grupo 1 de la clasificación de hipertensión pulmonar, es un trastorno
raro, muy complejo y progresivo, y que finalmente conduce a una muerte prematura. La HAP provoca importantes
inconvenientes físicos, sociales, laborales y emocionales entre los pacientes afectados y sus cuidadores. Se requiere,
en forma temprana, un diagnóstico y el inicio de un tratamiento lo más óptimo posible a fin de conseguir los mejores
resultados, tratando de detener el remodelamiento vascular y la falla cardíaca derecha; sin embargo, la presentación
clínica de la HAP es inespecífica y con frecuencia se asocia con otras comorbilidades, llevando a un retraso en el
diagnóstico o a un diagnóstico erróneo. En las últimas décadas, una mayor comprensión de la fisiopatología de la
HAP ha llevado a cambios en su definición, diagnóstico y tratamiento. Además, los registros contemporáneos de HAP
han mostrado mayores tasas de sobrevida entre los pacientes con HAP y han permitido el desarrollo de herramientas
de cálculo de riesgo que ahora se utilizan para impulsar objetivos terapéuticos. Hasta la fecha, se han desarrollado
múltiples tratamientos específicos para la HAP que se dirigen a las 3 vías que contribuyen a la patogenia de la
disfunción endotelial de la HAP (vías de la prostaciclina, de la endotelina y del óxido nítrico). Debido a que la HAP
se subdivide en 7 subgrupos, es esencial que los individuos se agrupen adecuadamente para optimizar la eficacia
del tratamiento y la prevención de complicación y mejorar su evolución. Dado que el deterioro de la calidad de vida
relacionada con la salud de la HAP es multifactorial, es importante que los pacientes se eduquen en la patología,
participen en el proceso de toma de decisiones clínicas y tengan acceso a una atención multidisciplinaria, lo que
mejorará la compliance con las indicaciones médicas. Existe evidencia convincente de que la detección de HAP en
poblaciones de alto riesgo permitirá un diagnóstico e intervención tempranas, ofreciendo una oportunidad prometedora
para mejorar los resultados de los pacientes. Sin embargo, los métodos de detección que se utilizan habitualmente en la práctica clínica tienen limitaciones y es posible que se requiera una combinación de herramientas o parámetros
para mejorar la sensibilidad y la selectividad de los programas actuales. La creación de algoritmos de detección para
pacientes con riesgo de HAP con esclerosis sistémica ha aumentado la velocidad y la especificidad del diagnóstico,
mejorando potencialmente la sobrevida, y aunque los costos de la detección siguen siendo significativos, ellos resultan
irrelevantes ante el alto costo del tratamiento de esta enfermedad en etapas avanzadas. Se requiere el desarrollo y la
validación de algoritmos de detección de HAP de otras etiologías. La detección de HAP en pacientes asintomáticos
en riesgo y el desarrollo de enfoques basados en la detección en pacientes sintomáticos, donde el diagnóstico rara
vez se considera, son necesarios para mejorar las tasas de detección y reducir el tiempo hasta el diagnóstico. Existe
una necesidad clara e insatisfecha de mejoras en el diagnóstico, caracterización y manejo de los pacientes con HAP.
La HAP es una enfermedad que progresa rápidamente, incluso en pacientes con síntomas leves, y la intervención
terapéutica oportuna es esencial para influir en el pronóstico a largo plazo de los pacientes con HAP.
Palabras clave: Hipertensión arterial pulmonar; Insuficiencia cardíaca derecha; Detección precoz; Diagnóstico temprano; Screening; Biomarcadores
Summary
The need for early detection of pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH), Group 1 of the pulmonary hypertension classification, is a rare, highly complex
and progressive disorder that ultimately leads to premature death. PAH causes significant physical, social, occupational,
and emotional inconveniences among affected patients and their caregivers. Early diagnosis and initiation of the most
optimal treatment possible are required in order to achieve the best results, trying to stop vascular remodeling and right
heart failure; however, the clinical presentation of PAH is nonspecific and is often associated with other comorbidities,
leading to delayed diagnosis or misdiagnosis. In recent decades, a greater understanding of the pathophysiology of
PAH has led to changes in its definition, diagnosis, and treatment. In addition, contemporary PAH registries have shown
higher survival rates among PAH patients and have allowed the development of risk calculation tools that are now used
to drive therapeutic targets. To date, multiple PAH-specific treatments have been developed that target all 3 pathways
that contribute to the pathogenesis of PAH endothelial dysfunction (prostacyclin, endothelin, and nitric oxide pathways).
Because PAH is subdivided into 7 subgroups, it is essential that individuals be properly grouped to optimize treatment
efficacy and complication prevention and improve outcome. Since the impairment of health-related quality of life in PAH
is multifactorial, it is important for patients to be educated in the pathology, participate in the clinical decision-making
process, and have access to multidisciplinary care, which will improve compliance with medical indications. There is
convincing evidence that screening for PAH in high-risk populations will enable earlier diagnosis and intervention, offering
a promising opportunity to improve patient outcomes. However, detection methods commonly used in clinical practice
have limitations and a combination of tools or parameters may be required to improve the sensitivity and selectivity of
current programs. The creation of detection algorithms for patients at risk of PAH with systemic sclerosis has increased the speed and specificity of diagnosis, potentially improving survival, and although the costs of detection remain significant,
they are irrelevant given the high cost of treatment of this disease in advanced stages. The development and validation
of detection algorithms for PAH of other aetiologies is required. Screening for PAH in asymptomatic patients at risk and
developing screening-based approaches in symptomatic patients, where diagnosis is rarely considered, are needed to
improve detection rates and reduce time to diagnosis. There is a clear and unmet need for improvements in the diagnosis,
characterization and management of patients with PAH. PAH is a rapidly progressing disease, even in patients with mild
symptoms, and timely therapeutic intervention is essential to influence the long-term prognosis of patients with PAH.
Keywords: Pulmonary arterial hypertension; Right heart failure; Early detection; Early diagnosis; Screening;
Biomarkers
Resumo
A necessidade da detecção precoce da hipertensão arterial pulmonar
A hipertensão arterial pulmonar (HAP), Grupo 1 da classificação da hipertensão pulmonar, é uma doença
rara, de alta complexidade e progressiva que acaba por levar à morte prematura. A HAP causa significativos
inconvenientes físicos, sociais, ocupacionais e emocionais entre os pacientes afetados e seus cuidadores. O
diagnóstico precoce e o início do tratamento mais otimizado possível são necessários para alcançar os melhores
resultados, tentando interromper o remodelamento vascular e a insuficiência cardíaca direita; no entanto, a
apresentação clínica da HAP é inespecífica e muitas vezes está associada a outras comorbidades, levando ao
diagnóstico tardio ou erro de diagnóstico. Nas últimas décadas, uma maior compreensão da fisiopatologia da
HAP levou a mudanças em sua definição, diagnóstico e tratamento. Além disso, os registros contemporâneos
de HAP mostraram maiores taxas de sobrevida entre os pacientes com HAP e permitiram o desenvolvimento
de ferramentas de cálculo de risco que agora são usadas para direcionar alvos terapêuticos. Até o momento,
vários tratamentos específicos para HAP foram desenvolvidos visando todas as 3 vias que contribuem para a
patogênese da disfunção endotelial da HAP (vias da prostaciclina, endotelina e óxido nítrico). Como a HAP
é subdividida em 7 subgrupos, é essencial que os indivíduos sejam agrupados adequadamente para otimizar
a eficácia do tratamento e a prevenção de complicações e melhorar o resultado. Como o comprometimento da
qualidade de vida relacionada à saúde na HAP é multifatorial, é importante que os pacientes sejam educados na
patologia, participem do processo de tomada de decisão clínica e tenham acesso a cuidados multidisciplinares,
o que melhorará a adesão às indicações médicas . Há evidências convincentes de que a detecção para HAP em
populações de alto risco permitirá diagnóstico e intervenção precoces, oferecendo uma oportunidade promissora
de melhorar os resultados dos pacientes. No entanto, os métodos de detecção comumente usados na prática clínica têm limitações e uma combinação de ferramentas ou parâmetros pode ser necessária para melhorar a
sensibilidade e a seletividade dos programas atuais. A criação de algoritmos de detecção para pacientes com
risco de HAP com esclerose sistêmica aumentou a velocidade e especificidade do diagnóstico, potencialmente melhorando a sobrevida e, embora os custos de detecção permaneçam significativos, são irrelevantes devido ao
alto custo do tratamento desta doença em estágio avançado. estágios. É necessário o desenvolvimento e validação
de algoritmos de detecção de HAP de outras etiologias. A detecção para HAP em pacientes assintomáticos em
risco e o desenvolvimento de abordagens baseadas em detecção em pacientes sintomáticos, onde o diagnóstico
raramente é considerado, são necessários para melhorar as taxas de detecção e reduzir o tempo até o diagnóstico.
Há uma necessidade clara e não atendida de melhorias no diagnóstico, caracterização e manejo dos pacientes
com HAP. A HAP é uma doença de progressão rápida, mesmo em pacientes com sintomas leves, e a intervenção
terapêutica oportuna é essencial para influenciar o prognóstico a longo prazo dos pacientes com HAP.
Palavras-chave: Hipertensão arterial pulmonar; Insuficiência cardíaca direita; Detecção precoce; Diagnóstico
precoce; Screening; Biomarcadores
Introducción
La hipertensión arterial pulmonar (HAP) es una enfermedad
rara y muy compleja, que afecta a los pulmones
y al corazón con repercusiones en todo el organismo. De
inicio insidioso y progresivo, conduce finalmente a una
muerte prematura, si no se detecta y trata precozmente
(sobrevida promedio sin tratamiento de 2 a 3 años)1-3.
La HAP conlleva importantes inconvenientes físicos,
sociales, laborales y emocionales entre los pacientes
afectados y sus cuidadores; por lo que es imprescindible
llegar a un diagnóstico temprano y un inicio de tratamiento
para tratar de obtener los mejores resultados,
tratando de detener el remodelamiento vascular y la
falla cardíaca derecha; sin embargo, la presentación
clínica de la HAP es inespecífica y con frecuencia se
asocia con otras comorbilidades, llevando a un retraso
en el diagnóstico o a un diagnóstico erróneo1-3.
La HAP es una enfermedad compleja y devastadora
que causa vasoconstricción progresiva y remodelación
vascular de las arterias pulmonares distales1,4-6. En el 6°
Simposio Mundial sobre Hipertensión Pulmonar (6th
World Symposium on Pulmonary Hypertension) de 2018
realizado en Niza, Francia, la hipertensión pulmonar
(HP) se clasificó en 5 grupos (Tabla 1)7, y la HAP
correspondió al Grupo 1 de dicha clasificación. La HP
describe un grupo de trastornos vasculares pulmonares
graves caracterizados por una presión arterial pulmonar
media (PAPm) elevada en reposo1,4.
Tabla 1. Clasificación clínica actualizada de hipertensión
pulmonar 2018
Debido a que la HAP se subdivide en 7 subgrupos
(Tabla 1)7, es esencial que los pacientes se agrupen
adecuadamente para poder obtener la mayor eficacia
del tratamiento a instituir, prevenir las complicaciones
y retardar, en lo posible, su evolución. Dado que el
deterioro de la calidad de vida relacionada con la salud
provocado por la HAP es multifactorial, es importante
que los pacientes participen en el proceso de toma de
decisiones clínicas y tengan acceso a una atención multidisciplinaria. La educación del paciente y su entorno
constituyen un pilar fundamental para mantener e incrementar
la compliance con las indicaciones médicas. Actualmente, no existe una cura y la mayoría de los pacientes
con HAP desarrollan con el tiempo una disfunción
del corazón derecho que conduce, indefectiblemente, a la muerte. Debido a la naturaleza progresiva de la HAP,
es crucial que la enfermedad sea diagnosticada tempranamente
con una clasificación precisa, donde el índice de sospecha constituye la clave para
la detección en los indicios de la
patología.
Los pacientes con HAP también
deben someterse a una evaluación
exhaustiva para determinar la gravedad
de la enfermedad y el riesgo
futuro, e idealmente tener acceso al
tratamiento en centros de atención
especializados1,4.
Las últimas dos décadas han estado
marcadas por avances significativos
conducidos por nuevas terapias
y una mejor comprensión de la
fisiopatología de la enfermedad8,
pero el tiempo desde el inicio de
los síntomas hasta el diagnóstico
se mantiene sin cambios en torno a
los 2 años3,9. Como resultado, el manejo
de la HAP está evolucionando
rápidamente.
A pesar de las pautas actualizadas
y los avances en el tratamiento,
el pronóstico a largo plazo para
los pacientes con HAP sigue siendo poco favorable,
estimándose que la mortalidad a 1 año se encuentra
entre el 8 y el 15 % en pacientes con HAP idiopática
(HAPI), familiar/hereditaria (HAPF/HAPH) o asociada
a anorexígeno10,11 y es de aproximadamente un 30% en
la HAP asociada con enfermedad del tejido conjuntivo10-12. Por ello, se hace necesario resaltar que sólo un
diagnóstico temprano y una intervención terapéutica
completa y precoz de la HAP pueden resultar en una
mejora en los resultados a largo plazo10,13-15.
Hasta hace poco tiempo, la HAP se consideraba una
enfermedad restringida a la circulación pulmonar. Sin embargo, existe una creciente evidencia de que
los pacientes con HAP también exhiben, además de
la remodelación vascular pulmonar, remodelación
vascular sistémica, alteraciones endocrinas y metabólicas,
inflamación vascular sistémica y disfunción del
musculo esquelético y respiratorio. Siendo estos cambios
morfológicos parte del cuadro clínico de la HAP,
pudiendo afectar negativamente el estado funcional de
estos pacientes. La HAP es una enfermedad pulmonar
con manifestaciones sistémicas que surgen de múltiples
factores, incluyendo respuestas endoteliales anormales,
desregulación metabólica e inflamación1. Aunque
las manifestaciones extrapulmonares en la HAP son a
menudo clínicamente sutiles, podrían tener un impacto
significativo en la evolución del paciente1.
La evidencia reciente demuestra que los programas de
detección de HAP en cuadros de esclerosis sistémicas
(ES) y otras manifestaciones de esclerosis pansistémicas
pueden identificar a los pacientes con formas más
leves de la enfermedad, lo que permite una intervención
terapéutica más temprana y una mejor supervivencia1,16 (Figura 1). Estos hallazgos brindan una base fundamental para respaldar la postura de realizar un diagnóstico
precoz y la posterior instauración de una terapéutica
temprana de la HAP, donde el pronóstico ha mostrado
una estrecha relación entre el grado funcional y la
supervivencia (Figura 2), recomendados por las guías
actuales10,11,13,14,17.

Figura 1. La evidencia sugiere que los pacientes con clase funcional I o II de la Organización
Mundial de la Salud tienen tasas de sobrevida a largo plazo (36 meses) significativamente
mejores que los pacientes con clases funcionales más altas, lo que proporciona una justificación para el diagnóstico y tratamiento más tempranos de la HAP.
Modificado de Humbert M y col.10

Figura 2. Representación esquemática del camino de la toma de consciencia para mejorar los resultados a largo plazo de la hipertensión
arterial pulmonar (HAP). Importancia de los registros y la vigilancia regular para la detección temprana de HAP. Screening en poblaciones
de riesgo.
Sin embargo, el diagnóstico oportuno de HAP es un
desafío por varias razones. Inicialmente, los síntomas
de la HAP suelen ser muy leves e inespecíficos, por
lo que es tan difícil identificar a los pacientes en esta
etapa (Figura 3). A medida que la enfermedad avanza,
se desarrollan síntomas inespecíficos como dificultad
para respirar o fatiga. En consecuencia, el diagnóstico
de HAP es un desafío. Otras condiciones como el
asma, la insuficiencia cardíaca (IC) crónica o incluso
la falta de actividad física o la depresión a menudo se
consideran antes que diagnosticar una HAP18.

Figura 3. Evolución de la historia clínica de los pacientes con HAP.
El diagnóstico temprano es un desafío y con frecuencia hay un retraso entre el inicio de los síntomas y el diagnóstico. Los pacientes con
frecuencia demoran la búsqueda de consejo médico después del inicio de los síntomas y tienen múltiples interacciones con profesionales
de atención primaria y especialistas una vez que lo hacen.
En pacientes con ES, las características músculo-esqueléticas coexistentes y la enfermedad pulmonar intersticial hacen que el diagnóstico de HAP sea un desafío aún mayor. Con frecuencia, esto da como resultado un retraso considerable entre el inicio de los síntomas y el diagnóstico de HAP. En promedio, el retraso entre el inicio de los síntomas y el diagnóstico es ≥2 años8, que no es sustancialmente diferente de la demora observada en el diagnóstico hace 25 años19. Esto significa que rara vez se sospecha una HAP hasta que ha alcanzado una etapa avanzada, y por lo tanto su pronóstico es malo18. Aunque las opciones de detección no invasivas se están expandiendo, un diagnóstico definitivo de HAP aún requiere un cateterismo cardíaco derecho (CCD)5,20-26. El propósito de esta actualización es concientizar a los profesionales de la salud sobre la detección y tratamiento precoz y completo de la HAP.
Diagnóstico clínico
El concepto fundamental en la valoración del diagnóstico
de la HAP es su precocidad y a partir de ello
iniciar su tratamiento tempranamente a fin de retardar
o evitar su progresión.
Detectar a los pacientes portadores de una HAP en una
etapa temprana no es tarea fácil, ya que su sintomatología
no es específica y sus manifestaciones clínicas son
incipientes en los estudios complementarios.
Básicamente, las etapas diagnósticas de la HAP son dos:
una etapa de detección y una etapa de caracterización
(Tabla 2)27.
Tabla 2. Etapas del diagnóstico de la hipertensión arterial pulmonar.

La etapa de detección está relacionada con el diagnóstico
de la HAP. La etapa de caracterización se basa en
definir el contexto clínico de cada paciente que presenta
HAP, es decir, cuál es el cuadro clínico acompañante. En base a ello, también se deben analizar los diferentes
pronósticos.
Si bien se puede realizar el diagnóstico de HAP por
exclusión, habitualmente cuando el paciente llega a la
consulta, en general, se encuentra en la etapa final de su
enfermedad; lo cual no nos posibilita detener o retardar
la progresión de la enfermedad en una fase temprana y
así tomar una actitud terapéutica precoz que mejore la
supervivencia y la calidad de vida de estos pacientes.
Actualmente contamos con una terapéutica mucho más
específica respecto a los mecanismos fisiopatológicos
de la HAP. Es por ello que tenemos muy buenas expectativas
y avanzadas investigaciones que dan evidencia de que nos encontramos en las etapas iniciales de tratamientos
cada vez más efectivos de esta enfermedad
tan lamentable.
En el registro francés de Humbert y col.8, entre 674 pacientes,
se reportó que el 75% de los pacientes llegan a la consulta
en clase funcional (CF) III-IV según la Organización
Mundial de la Salud (OMS) -basada en la clasificación
formulada por New York Heart Association (NYHA) para
IC izquierda y posteriormente adaptada para la HP por la
OMS-, y que la mayor incidencia de la enfermedad se daba
en mujeres con una edad media de 50 años.
Por otro lado, los síntomas no son patognomónicos de
esta patología (Tabla 3)19: disnea, fatiga, dolor de pecho,
síncope o pre-síncope y edema. Además, no estamos
acostumbrados a su diagnóstico precoz, y entonces la
subdiagnosticamos.
Tabla 3. Síntomas de la hipertensión arterial pulmonar2

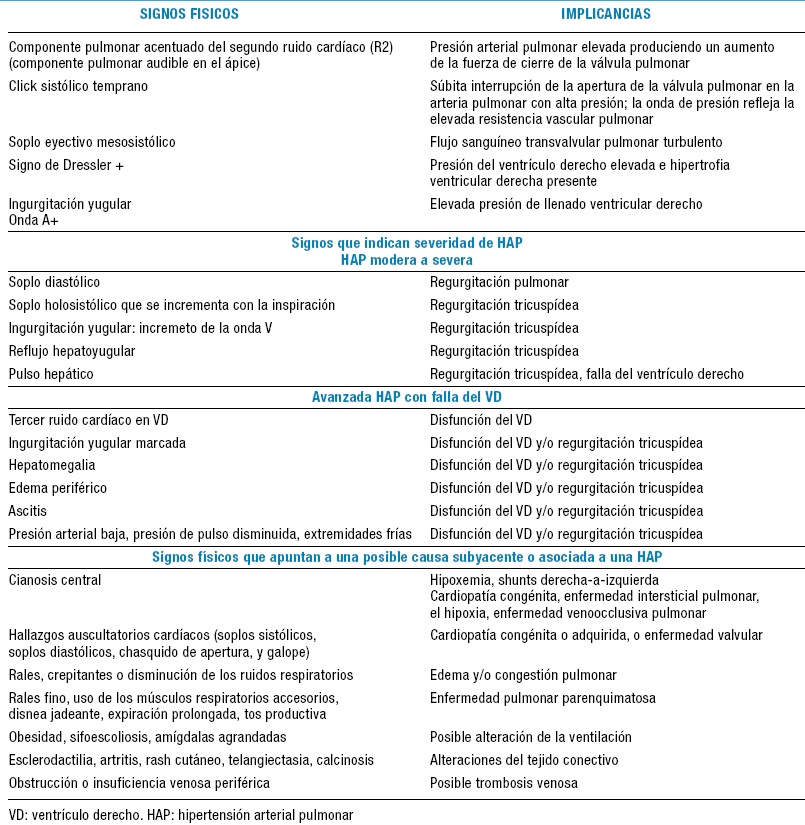
Recién en las etapas más avanzadas se empieza a sospechar de ella, pues la presentación es más florida y discapacitante, y es cuando el paciente se hace refractario y no responde a las terapéuticas específicas (Tabla 4)28. En la detección, entonces, puede ayudarnos el hecho de encontrar algunas manifestaciones en el examen físico: el 2° ruido cardíaco pulmonar aumentado, un soplo sistólico de insuficiencia tricuspídea, el soplo diastólico de Graham-Still de insuficiencia pulmonar; un 3° ruido derecho, regurgitación yugular, manifestaciones de ascitis, distención abdominal, edema de miembros inferiores, o extremidades frías. Cada uno de estos signos y síntomas no son absolutamente específicos29,30, pero deben hacernos sospechar la posibilidad de HP para indicar los estudios que nos ayudarán a un diagnóstico más preciso.
Tabla 4. Signos clínicos que indican HAP.
En la telerradiografía (Rx) de tórax, podemos encontrar agrandamiento del ventrículo derecho (VD), de la aurícula derecha (AD), las arterias pulmonares dilatadas, a veces con un stop, que impide ver la vasculatura periférica (Figura 4)31. El electrocardiograma (ECG) no parece ser una buena herramienta para el screening de HAP. Se postula que tiene una sensibilidad de aproximadamente el 55%, y una especificidad del 70%, pero los hallazgos más significativos son, sin duda, la hipertrofia ventricular derecha con signos de sobrecarga de presión, el agrandamiento de la AD, y la presencia de un eje cardíaco desviado hacia la derecha (Figura 5)32.
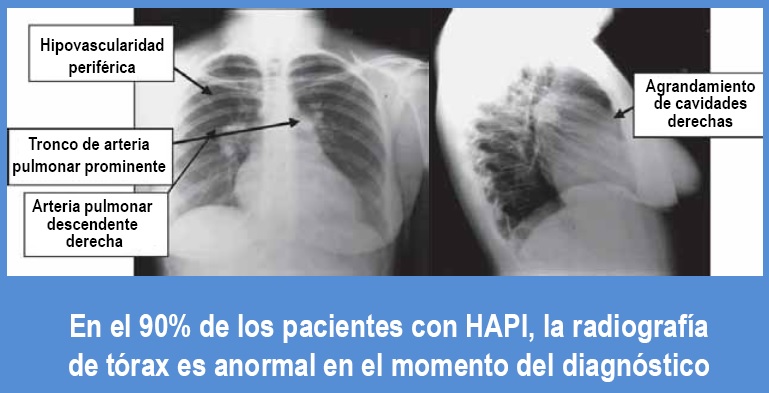
Figura 4. Telerradiografía de tórax en pacientes con hipertensión arterial pulmonar idiopática (HAPI). Los hallazgos incluyen dilatación
de la arteria pulmonar central, que contrasta con la “poda” (pérdida) de los vasos sanguíneos periféricos. La aurícula derecha y el
agrandamiento del ventrículo derecho pueden verse en casos más avanzados. La radiografía de tórax permite descartar razonablemente
enfermedades pulmonares moderadas a severas asociadas o hipertensión venosa pulmonar debida a cardiopatía izquierda. En general, el
grado de hipertensión pulmonar en cualquier paciente no se correlaciona con la extensión de las anomalías radiográficas.
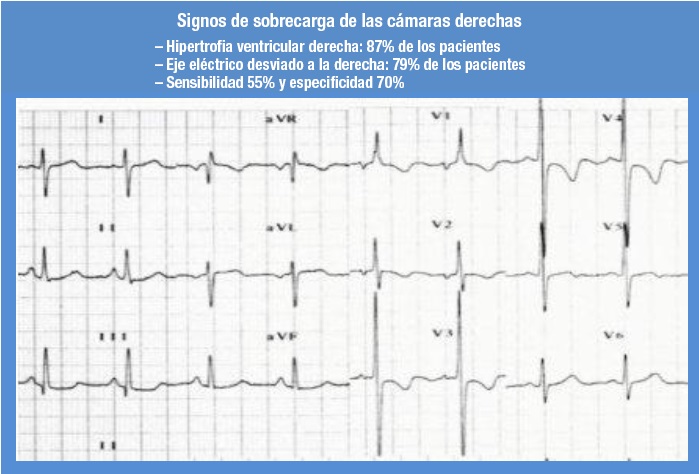
Figura 5. Los hallazgos electrocardiográficos de la hipertensión pulmonar (HP) no son específicos ni sensibles y su ausencia no descarta
la presencia de HP. Las presiones pulmonares elevadas en la HP provocan hipertrofia ventricular derecha (HVD) y agrandamiento de la
aurícula derecha que se evidencian en el trazado electrocardiográfico por desviación del eje a la derecha, rotación horaria, patrón de HVD
con sobrecargas de presión del ventrículo derecho y P pulmonale.
Evidentemente, estos signos en el ECG y la Rx de tórax como consecuencia de la progresión de la HAP necesitan un tiempo para manifestarse. Y cuando llegamos a este diagnóstico, utilizando sólo estos métodos, los 2,8 años de sobrevida original promedio de la HAP se han acortado, pues no se ha realizado un diagnóstico precoz y se ha perdido un tiempo valiosísimo para el paciente y su mejora evolutiva, si hubiera recibido el tratamiento específico en forma precoz (Tabla 5)19,33,34. Es así que debemos considerar al ecocardiograma Doppler transtorácico en la detección de esta patología, que nos permitirá, inicialmente, antes de realizar estudios más invasivos, detectar que el paciente padece una HAP35. En principio, con todos estos hallazgos, es posible diagnosticar HAP.
Tabla 5 . Historia natural de la hipertensión arterial pulmonar

El objetivo es detectar mediante un examen muy sencillo el diagnóstico precoz de estas patología y, desde ya, permitir un tratamiento más eficiente, y así poder mejorar la evolución de un paciente que aún se encuentra en CF I o II, cuando sus presiones pulmonares no son tan elevadas, y quizás sin muchas manifestaciones clínicas de IC derecha (Tabla 6).
Tabla 6. Estrategia diagnóstica en HAP4.

El diagnóstico de HAP se define con el CCD por una PAPm ≥20 mm Hg con una presión capilar pulmonar en cuña o enclavamiento -wedge- (PCP) ≤15 mm Hg7. Los criterios de diagnóstico adicionales pueden incluir un gasto cardíaco normal o reducido o una resistencia vascular pulmonar (RVP) >3 unidades de Wood (Tabla 7)7.
Tabla 7. Definiciones hemodinámicas de hipertensión pulmonar.

La RVP aumenta progresivamente, tanto en la etapa asintomática como en la sintomática. Pero sucede en forma diferente con la presión arterial pulmonar; pues ésta también sube, hasta que en un momento determinado empieza a caer, ante la presencia de falla ventricular derecha. Es muy importante detectar el compromiso de la función ventricular derecha, porque cuando ello sucede, probablemente, estemos en presencia de una incipiente fase de IC derecha que llevará a la claudicación del VD, ya que los pacientes con signos de IC derecha [hepatomegalia, edemas de miembros inferiores, trastornos de coagulación, agotamiento, aspecto azulado (cianosis), y presencia de cuadros sincopales] presentan una evolución desfavorable (Figura 6).

Figura 6. Representación esquemática de la evolución de la hipertensión arterial pulmonar con deterioro progresivo final por claudicación
del ventrículo derecho.
Lo mismo ocurre con el volumen minuto (VM) cardíaco
y la presión de la AD. El VM en la primera etapa se
mantiene, pero luego va disminuyendo, y la presión de
la AD empieza a aumentar, manifestando los signos de
IC derecha.
En conclusión, los síntomas son relativamente
ambiguos y solamente en las fases más avanzadas
se hacen más evidentes, indicando la presencia de
IC derecha. Pero, estos momentos son tardíos para
conseguir una terapia eficaz y revertir el cuadro clínico.
El mensaje es detectar los síntomas, o buscar
elementos clínicos que puedan hacer sospechar lo
más precozmente posible esta enfermedad, para tratar
que los mecanismos fisiopatológicos que agravan la
HAP puedan ser detenidos, al menos parcialmente,
para lograr una mayor sobrevida y mejorar la calidad
de vida del paciente.
Una vez realizado el diagnóstico de HAP, pasamos a
la etapa de caracterización de la misma, y para ello, es
necesario realizar estudios esenciales (que no deben
faltar en ninguno de los pacientes), y estudios ocasionales (aquellos a los que podríamos recurrir en algunos
pacientes y en determinadas condiciones) (Tabla 2)36-39.
Detección de la HAP
La detección se puede definir como la sistematización
de una o varias pruebas en pacientes en riesgo para
detectar enfermedades en una etapa preclínica3. El
objetivo de la detección precoz es identificar a los
pacientes con síntomas leves, así como a aquellos con
enfermedad preclínica, con la finalidad de prevenir o
retrasar la progresión de la enfermedad mediante un
tratamiento precoz9. Los programas de detección juegan
un papel importante en ciertas poblaciones en riesgo de
desarrollar una HAP y pueden permitir que los pacientes
sean identificados en una etapa más temprana que en
la práctica clínica habitual40. Se reconoce que ciertas
condiciones médicas y susceptibilidades genéticas
predisponen a una persona al desarrollo de HAP. Estos
incluyen, pero no se limitan a, mutaciones del receptor
de proteína morfogenética ósea tipo 2 (BMPR2), un
familiar de primer grado con mutación BMPR2, infección
por VIH (virus de la inmunodeficiencia humana),
enfermedad cardíaca congénita con derivación, ES,
enfermedad de células falciformes y embolia pulmonar
aguda reciente2,41.
Pueden transcurrir años entre el inicio de los síntomas
y el diagnóstico de HAP, por lo que es valioso considerar
la adopción de enfoques de detección de HAP
en pacientes asintomáticos (de riesgo) y sintomáticos
(síntomas y signos inespecíficos), donde la HAP rara
vez se considera en el diagnóstico diferencial inicial.
Idealmente, una prueba de detección debería tener una alta sensibilidad y especificidad, ser reproducible, no invasiva,
económica, de fácil acceso y poder realizarse en
entornos donde los resultados se puedan aplicar con más
pruebas de confirmación o un tratamiento específico.
Sin embargo, aun hay muchos interrogantes a la hora
de conceptualizar un programa de detección para maximizar
la relación riesgo-beneficio para cada paciente.
Por ejemplo, la HAP afecta al 7-12% de los pacientes
con ES40,42 y es responsable de casi el 30% de las
muertes relacionadas con ella12. El valor de la detección
de HAP en pacientes con ES ha sido destacado
por Humbert y col.16 en un estudio prospectivo de 16
pacientes con ES cuya HAP se detectó en un programa
de screening/detección precoz, donde se compararon
con 16 pacientes con ES cuya HAP se diagnosticó durante
la práctica clínica habitual. En el momento del
diagnóstico de HAP, los pacientes detectados mediante
screening tenían enfermedad vascular pulmonar menos
avanzada que los pacientes identificados en la práctica
diaria habitual, como lo demuestra su PAPm más baja,
una RVP más baja y un gasto cardíaco más alto16. En
el momento del diagnóstico, el 6% de los pacientes
detectados mediante screening estaban en CF I según
OMS, el 44% en CF II, el 50% en CF III y ninguno
en CF IV (Figura 7). Estos resultados contrastan fuertemente
con los de los pacientes diagnosticados en la
práctica habitual, en los que la mayoría de los pacientes
ya estaban en CF III o IV (OMS) en el momento del
diagnóstico (69% y 18,5%; respectivamente) (Figura
7)16. Además, ninguno de los pacientes diagnosticados
de HAP durante la práctica habitual se encontraba en
el CF I en el momento del diagnóstico. La detección
temprana de HAP en pacientes con ES y la intervención
terapéutica temprana pueden tener un impacto importante en los resultados a largo plazo. Los pacientes con
HAP y ES identificados en el programa de detección
tuvieron estimaciones de sobrevida según las curvas
de Kaplan-Meier significativamente más altas a los
8 años que los pacientes identificados por la práctica
diaria de rutina (64% versus 17%; p=0,0037). Estos
datos resaltan la importancia de la detección temprana
y presumiblemente la intervención temprana en los
resultados a largo plazo de los pacientes con HAP y ES.
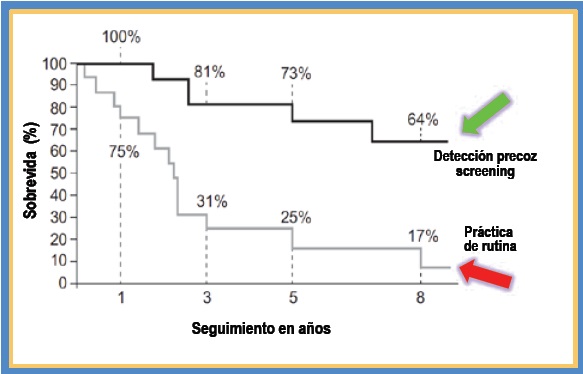
Figura 7. La evidencia sugiere que los pacientes con clase funcional de la Organización Mundial de la Salud detectados como parte de un
programa de screening tienen tasas de sobrevida a largo plazo significativamente mejores que los pacientes detectados durante la práctica
habitual, proporcionando una justificación para el diagnóstico y tratamiento más tempranos de la HAP.
Modificado de Humbert M y col.16
La IC derecha como resultado de HAP es una de las principales causas de muerte en esta población de pacientes,
representando el 26% de las muertes12, y la ES
asociada a la HAP representa el 15-20% de todas las
formas de HAP8 con un 30% de mortalidad a 1 año43,44.
Existe un interés significativo en desarrollar herramientas
de detección mejoradas utilizando una variedad
de enfoques que incluyen biomarcadores sanguíneos,
imágenes, pruebas de esfuerzo45 y datos de utilización
de recursos de atención médica46,47 para mejorar la detección
de HAP y disminuir el tiempo desde el primer
síntoma hasta el diagnóstico3. El screening (toma de
conciencia de la existencia) de HAP en una población
de riesgo de pacientes con ES permite el diagnóstico
temprano y, por tanto, el inicio del tratamiento en presencia
de una forma más leve de la enfermedad16. Esto
resulta en un aumento significativo en la sobrevida,
pero es importante reconocer los sesgos de tiempo de
anticipación y duración introducidos por el enfoque del
estudio de detección16, y el tratamiento más temprano
sigue siendo un objetivo importante para la HAP. Con
este fin, el desarrollo de algoritmos como el DETECT,
que mejoran el diagnóstico basado en síntomas y
ecocardiografía transtorácica en un subconjunto de
pacientes con ES con etapas tempranas de HAP48,49,
representa una herramienta prometedora2.
El algoritmo DETECT incluye valores para los biomarcadores
circulantes N-terminal pro-péptido natriurético
cerebral (NT-proBNP) y ácido úrico, lo que demuestra
el potencial de los biomarcadores para apoyar la detección
temprana de HAP en pacientes con ES, clasificando
a los pacientes con ES en aquellos con y sin HAP.
La detección de HAP no está exenta de desafíos, y a pesar
de las recomendaciones de los grupos de consenso y
las pautas para la detección anual en personas en riesgo,
existe evidencia limitada para respaldar esta directiva.
El mayor problema con cualquier prueba de detección
es cómo calibrar esta herramienta, pues utilizando una
prueba de detección con una alta especificidad garantizará
que la mayoría de los pacientes identificados tengan
HAP. Sin embargo, la alta especificidad a menudo se
produce a expensas de la baja sensibilidad, por lo que es
probable que se pasen por alto más pacientes con HAP.
Si la prueba de detección se calibrara para una especificidad
más baja, se encontrarán más personas con
HAP, pero a expensas de un mayor número de falsos
positivos, ya que más pacientes se someterán a un CCD
innecesario.
Lograr un equilibrio adecuado entre el número de
referencias de CCD y la tasa de diagnósticos de HAP
equivocados no sólo es un conflicto clínico, sino también
financiero y riesgoso, para todos los involucrados.
Se han desarrollado otros medios de detección, complementarios
a la ecocardiografía, como por ejemplo, los
que incluyen un sistema de puntuación simple, basado
en observaciones clínicas de rutina50 y una fórmula de
estratificación de riesgo para determinar la función pulmonar51,
ayudando a mejorar la selección de pacientes
para el CCD.
Lamentablemente, los programas de detección que
utilizan únicamente ecocardiografía Doppler tienen
limitaciones. Hachulla y col.52 demostraron que la ecocardiografía
es menos confiable como indicador de HAP
en pacientes con un velocidad máxima de regurgitación
tricuspídea (VmRT) entre 2,5 y 2,8 mseg-1 (pacientes
con enfermedad menos severa)40,52. Otro estudio que investigó
la sensibilidad y especificidad de la VmRT para
detectar HP asintomática en adultos con enfermedad
de células falciformes, demostró un valor predictivo
positivo de 25% a 64%; pero, el número de diagnósticos
falsos negativos fue del 42%53. Por lo tanto, el uso de la
ecocardiografía sola puede ser insuficiente para detectar
HP, necesitándose el uso de herramientas de detección
adicionales, fundamentalmente en aquellos pacientes
en los cuales el riesgo de un error diagnóstico puede
comprometer su supervivencia54.
Otro tema a considerar es el rendimiento de cualquier
prueba de detección en poblaciones de pacientes dispares
y, además, las herramientas de detección funcionan
de manera diferente en varias poblaciones, por ejemplo,
la capacidad de difusión del pulmón para el monóxido
de carbono (DLCO) reducida que presenta la HAP en
una ES parece particularmente diagnóstica, pero no
tiene relevancia en la HAP idiopática. En el estudio de
Parent y col. de pacientes con HP asociada a la enfermedad
de células falciformes, la prueba de la marcha
de 6 minutos (PM6M) pareció tener un valor discriminatorio,
mientras que esta herramienta no demostró ser
relevante en las poblaciones con ES53. Obviamente, la
ecocardiografía es más precisa en pacientes con enfermedad
avanzada.
La sensibilidad y especificidad subóptimas de las estrategias
de detección actuales para poblaciones de alto
riesgo enfatiza la necesidad de enfoques alternativos
para mejorar la selección de pacientes para derivación
para CCD y diagnóstico definitivo. Las herramientas
de detección actualmente disponibles incluyen ecocardiografía
Doppler, evaluación de la disnea, pruebas de
función pulmonar y los biomarcadores séricos (NTproBNP)
50,51,54. Estas pruebas detectan HAP utilizando
diferentes mecanismos biológicos. Actualmente, no
está claro si una combinación de dos o más de estas
herramientas puede mejorar la especificidad o la sensibilidad
de los análisis de detección, sugiriéndose que
se podría utilizar una combinación de ecocardiografía
y análisis de transferencia de gases para enriquecer la
población de detección.
Los biomarcadores también pueden tener utilidad en la
detección de HAP. Los hallazgos de Allanore y col.55 sugieren que el uso de una combinación de pruebas de
función pulmonar y la medición de NT-proBNP puede
ser un medio útil para evaluar a los pacientes en cuanto
a la presencia o ausencia de HAP. Seguramente, la
utilización de una combinación de marcadores clínicos,
serológicos, ecocardiográficos, etc. nos ayudará en la
detección de una HAP en los pacientes.
A pesar de las limitaciones de los métodos de estudio
para detectar precozmente una HAP48; actualmente, la
clave está en sensibilizar a los profesionales de atención
primaria para lograr la derivación oportuna a los centros
especializados ante una sospecha de HAP.
Poblaciones de riesgo
La realización de una detección programada no es
factible en todos los pacientes asintomáticos en riesgo,
por lo que es esencial que se seleccionen las poblaciones
apropiadas. Las recomendaciones basadas en
la evidencia para la detección y el diagnóstico de la
HAP actualmente solo están disponibles para la HAP
asociada con enfermedades del tejido conectivo (ETC)56 y establecen que los pacientes con ES, ETC mixta u
otras ETC con características de esclerodermia deben
someterse a detección de HAP. Sin embargo, la prevalencia
de HAP en pacientes con ETC distintos de la ES
puede ser demasiado baja para justificar la detección a
gran escala de pacientes asintomáticos, utilizando los
enfoques actuales. En el lupus eritematoso sistémico,
donde se cree que la prevalencia de HAP es de aproximadamente
el 1% y, por lo tanto, considerablemente
más baja que la ES, actualmente se están investigando
los beneficios potenciales de la detección57.
Otros grupos de pacientes con una alta prevalencia de
HAP incluyen individuos con cardiopatía congénita,
hipertensión portal, infección por VIH o predisposición
genética a la HAP7.
Las mutaciones heterocigóticas del gen que codifica el BMPR2 representan aproximadamente el 80% de los
casos de HAPF/HAPH y entre el 10 y el 20% de los
casos esporádicos2,20,58,59; los pacientes con mutaciones
en BMPR2 son más jóvenes y tienen una enfermedad
más grave en el momento del diagnóstico y tienen un
mayor riesgo de muerte o de trasplante pulmonar en
comparación con aquellos sin mutaciones en BMPR260-62. Por lo tanto, la detección genética es potencialmente
un beneficio considerable, aunque se debe considerar
el impacto emocional en los pacientes y los miembros
de la familia que tienen mutaciones asociadas con la
HAP. Las conclusiones del estudio DELPHI-2, que
representa el primer estudio que examina sistemáticamente
la detección de HAP en portadores asintomáticos
de la mutación BMPR2, arrojaron que los portadores
asintomáticos de la mutación BMPR2 tienen un riesgo
significativo de desarrollar HAP incidente que va desde
el 0,99% anual en los hombres hasta el 3,5 % anual en
las mujeres. Los datos de DELPHI-2 indican que se
puede implementar en la práctica clínica un programa
de detección multimodal seguido de un seguimiento
cuidadoso anual para permitir la detección y el tratamiento
tempranos de HAP en portadores de la mutación BMPR2, necesitándose estudios multicéntricos internacionales
para confirmar que los programas de screening
multimodal perfeccionados con un seguimiento periódico
permiten la detección precoz de la HAP 63 .
Futuros estudios familiares en pacientes con HAPF/
HAPH confirmados, como el estudio NAIAD (National
Cohort Study of Idiopathic and Heritable Pulmonary
Arterial Hypertension) del Reino Unido sobre HAPI y
HAPH (NCT01907295), podrían ayudar a establecer
el riesgo de desarrollar HAP según el genotipo64. Sin
embargo, tales estudios están restringidos por un número
relativamente pequeño de pacientes y el tiempo
requerido para acumular datos. Todo indica que se
requieren más estudios para determinar la viabilidad de
la identificación de éste y otros rasgos genéticos para
predecir el desarrollo de HAP.
De igual manera, existe un bajo porcentaje de pacientes
asintomáticos con hipertensión portal (2% - 6%)65 o VIH
(0,5%)66 que desarrollan HAP, hace que la detección a
gran escala no sea aconsejable; sin embargo, los riesgos
asociados a la cirugía en presencia de una HAP hacen
lógico ofrecer la detección de la HAP en pacientes con
hipertensión portal derivados para trasplante hepático.
Actualmente no existen guías específicas basadas en
la evidencia para la detección de HAP en cardiopatías
congénitas, aunque estos pacientes deben ser tratados
en centros especializados y evaluados en pacientes de
riesgo.
Estudios utilizados para detectar HAP
Rx de tórax, ECG y ecocardiografía
Como ya se han mencionado, los estudios de rutina de
la práctica diaria, como la Rx de tórax, el ECG y el
ecocardiograma, detectan la HP y su repercusión en el
organismo y especialmente, en el corazón y los pulmones2.
Obviamente, ésta es una carrera contrarreloj, y los
signos son más evidentes cuanto más haya progresado
la enfermedad.
La Rx de tórax tiene un limitado beneficio, no obstante,
en el 90% de los pacientes con HAP la radiografía
torácica es anormal en el momento de realizar el diagnóstico20,33,67.
En general, el grado de la HP en cualquier paciente
no está en correlación con el de las anomalías radiográficas.
Los signos radiológicos torácicos han sido
evaluados para hipertensión pulmonar, encontrándose
baja sensibilidad y buena especificidad2.
Se ha implementado en el Japón un sistema de detección
masiva basado en electrocardiografía para enfermeda des cardíacas pediátricas generales. Entre 2005 y 2012,
este screening detectó una subpoblación de pacientes
pediátricos con HAPI y HPAH asociada con hemodinámica
pulmonar avanzada en reposo en sujetos sin
IC derecha incuestionable68. Aunque la prueba de detección
no logró identificar la HAP en etapa temprana,
sí mejoró la detección de la enfermedad. En pacientes
con ES examinados para HAP en el estudio DETECT,
la presencia de desviación del eje a la derecha ayudó a
discriminar entre pacientes con y sin HAP y se incorporó
en el algoritmo de detección48.
Otro método utilizado en la práctica diaria es la ecocardiografía
transtorácica con Doppler (ETT) que se
recomienda siempre que se sospeche HAP2, incluso
un estudio reciente que incluyó a 313 492 pacientes
sometidos a ecocardiografía demostró que la ETT se
usa ampliamente y tiene el potencial para identificar
pacientes a nivel poblacional. Es importante destacar
que cuando existen leves elevaciones de la presión de
la arteria pulmonar sistólica (PAPS), éstas se asociaron
con un mal resultado69. La velocidad de regurgitación
tricuspídea (VRT) es un parámetro importante de la
ETT, que permite estimar la PAPS. Aunque los valores
más altos de VRT se asocian con una mayor probabilidad
de HAP, las estimaciones de PAPS pueden ser
inexactas70.
Otros parámetros ecocardiográficos asociados con HAP
incluyen: mediciones de los ventrículos, alteraciones
del septum interventricular, de la arteria pulmonar,
aumento en el diámetro de la vena cava inferior o del
área de la AD, y otros2,71-73. Basado en las evidencias
del estudio DETECT48, que incluyó una amplia gama de
variables ecocardiográficas en su modelo de detección,
se sugirió que la VRT y el área de la AD fueran parámetros
ecocardiográficos clave para evaluar pacientes
con ES con el fin de su eventual derivación al CCD.
Pruebas de función pulmonar
La mayoría de los pacientes con HAP tienen una DLCO
disminuida2. Se observan con frecuencia reducciones
significativas en la DLCO en pacientes asintomáticos
que posteriormente se les diagnostica HAP y ES, considerándose
un predictor sólido de la enfermedad40.
En particular, una DLCO <60% puede usarse para enriquecer
a una población con mayor riesgo de HAP48.
Aproximadamente el 75% de los pacientes con HAPI,
en particular los pacientes mayores o aquellos con una
mayor exposición al tabaco, tienen una DLCO reducida38,74.
Sin embargo, una DLCO normal no excluye un
diagnóstico, particularmente en pacientes con HAPF/
HAPH, donde la DLCO puede estar consevada2.
Biomarcadores sanguíneos
Los estudios de biomarcadores para detectar HAP se
encuentran en una etapa temprana de desarrollo y la
mayoría de los estudios que destacan un papel potencial
para los biomarcadores sanguíneos se han realizado en
poblaciones enriquecidas de pacientes con diagnósticos
de HAP sospechosos o confirmados45,75-77. De los biomarcadores
sanguíneos propuestos actualmente, solo el
péptido natriurético cerebral (BNP), el NT-pro-BNP y
el ácido úrico sérico están incluidos en los algoritmos
de detección utilizados actualmente48. La HP provoca
un aumento en el estrés de la pared miocárdica que
resulta en la liberación de NT-pro-BNP (precursor
inactivo de BNP) por parte de los cardiomiocitos78,79.
Se ha demostrado que la concentración sanguínea de
este péptido es un biomarcador útil para la HAP2,48,80,81 particularmente en la HAP con ES, aunque es posible
que no identifique a los pacientes con enfermedad leve48.
Los niveles sanguíneos elevados de NT-pro-BNP también
pueden verse en pacientes con afecciones como
disfunción ventricular izquierda e insuficiencia renal
avanzada, por lo que se necesita un minucioso cuidado
para eliminar cualquier otra fuente potencial de la anomalía
antes de realizar más evaluaciones. Un pequeño
número de estudios ha demostrado que los niveles de
ácido úrico plasmático están elevados en pacientes con
HAP y reflejan la gravedad de la enfermedad82,83. El
ácido úrico plasmático es el producto final de la degradación
de las purinas y se cree que los niveles elevados
reflejan una alteración del metabolismo oxidativo que
se produce como consecuencia de la isquemia tisular84.
En el estudio DETECT, los niveles elevados fueron
predictivos de HAP48.
El valor diagnóstico y pronóstico de los biomarcadores
sanguíneos en sujetos asintomáticos o poblaciones específicas
en riesgo necesita más investigación, ya que
tienen el potencial de desempeñar un papel importante
en los programas de detección, dada la simplicidad de
los análisis de sangre.
Necesidades futuras
El desarrollo y la implementación de algoritmos de
detección de HAP para pacientes con ES ha demostrado
que los pacientes con HAP pueden ser diagnosticados
con una enfermedad hemodinámica menos grave3,9,48,56,
requiriéndose más investigación para identificar a los
pacientes con ES que tienen bajo riesgo de desarrollar
HAP. El éxito de los algoritmos de detección en pacientes
con ES es alentador y subraya la importancia
de desarrollar algoritmos que sean adecuados para su
uso en otros pacientes con riesgo de desarrollar HAP.
Sin embargo, una gran proporción de pacientes con
HAP no pertenecen a una población “en riesgo” y la
identificación de estos pacientes con síntomas leves
sigue siendo un desafío. Actualmente, el diagnóstico
oportuno en estos pacientes se basa en un enfoque sistemático
para la investigación de estos pacientes en los
entornos de atención primaria y secundaria. El único
estudio que investigó el impacto de una herramienta
de detección “tradicional” en la detección de HAP en
una población general no seleccionada es un estudio nacional reciente de detección masiva basada en electrocardiografía
en escuelas japonesas68. La detección
sistematizada basada en la electrocardiografía para la
detección precoz de diversas enfermedades cardiovasculares
ha sido obligatorio en las escuelas japonesas
desde 1995, pero Sawada y col. fueron los primeros en
investigar el impacto de este sistema establecido en la
detección de la HAP.
Esto indica que este programa de detección detectó
una subpoblación de pacientes con HAPI/HPAH, pero
no logró una detección temprana “verdadera”, ya que
la PAP elevada es un evento relativamente tardío en el
desarrollo de HAP.
Otras alternativas para detectar HAP
Nuevos biomarcadores
El uso de biomarcadores plasmáticos para detectar
HAP es atractivo, ya que el muestreo de sangre es
sencillo y se puede implementar fácilmente a nivel de
población. El enfoque actual de los biomarcadores en
sangre se ha basado principalmente en la identificación
de biomarcadores que reflejan el impacto del proceso
de la enfermedad en la función cardíaca en lugar de
centrarse en biomarcadores que reflejan anomalías de
la vasculatura pulmonar. Los niveles elevados de NTpro-BNP sólo se observan en pacientes con HAP e IC,
mientras que los enfoques proteómicos, metabolómicos
y de microARN para la identificación de biomarcadores
pueden tener el potencial de ayudar a un diagnóstico
más temprano.
Los estudios proteómicos están adquiriendo en los
últimos años una gran relevancia, fundamentalmente
en lo que hace referencia a su aplicación a la patología
humana. Con este fin se están realizando un gran número
de estudios en plasma humano, tejidos y diversos
líquidos biológicos. La utilidad práctica de los resultados
obtenidos con la proteómica en relación con la salud
es muy importante. El descubrimiento de marcadores
proteicos de afecciones como las cardiovasculares,
neurológicas, oncológicas, metabólicas, entre otras,
tiene una aplicación clínica inmediata en el diagnóstico,
el seguimiento y el tratamiento de estas enfermedades.
La proteómica es una tecnología en desarrollo que
investiga la estructura y función del conjunto de proteínas
que conforman el proteoma. El término proteoma
apareció en 1994 como un equivalente lingüístico del
concepto de genoma85.
El interés de la proteómica se centra en el conocimiento
del conjunto de las interacciones entre proteínas para
constituir la red de interacciones, que caracteriza el
funcionamiento de los organismos vivos. En otras
palabras, la proteómica es el estudio del proteoma86.
Una de las diferencias fundamentales entre la proteómica
y el estudio clásico de las proteínas es su carácter
global. No se centra en el estudio de una determinada
proteína, sino que consiste en un estudio de aproximación
al funcionamiento del conjunto de proteínas85.
Actualmente, la investigación del proteoma se centra,
principalmente, en la puesta a punto de métodos exactos
y relativamente rápidos para identificar y caracterizar
el conjunto de proteínas85.
Entre las tecnologías clásicas que se utilizan, se encuentran
la electroforesis bidimensional y la espectrometría
de masas. Otra técnica más novedosa que ya
ha proporcionado buenos resultados son los chips de
proteínas que, al igual que los chips de ADN, permiten
el análisis simultáneo de miles de proteínas. Además,
gracias a la bioinformática, existen programas capaces
de determinar las reacciones metabólicas que tiene
una determinada proteína con el resto del proteoma86.
Los microARN (pequeñas moléculas de ARN no
codificantes que regulan la expresión génica a nivel
post-transcripcional de las proteínas) son estables y
se detectan fácilmente en plasma y varios estudios
han demostrado que están desregulados en pacientes
con HAP87,88 con niveles circulantes que predicen la
sobrevida, por ejemplo, en los casos de reducción de
microRNA-15089 y regulación de microRNA-140-5p90.
Los análisis proteómicos y metabolómicos también
identificaron una combinación de nueve proteínas circulantes
asociadas con un alto riesgo de mortalidad,
independientemente de otras evaluaciones clínicas79,91,
siendo metabolitos pronósticos para HAP, con ARN
de transferencia modificado y bioenergética alterada
relacionada con sobrevida92. Sería revelador explorar
la asociación de más marcadores sanguíneos a los algoritmos
de detección existentes, como DETECT48, y
estrategias de estratificación de riesgo, como la calculadora
de puntaje de riesgo REVEAL80,93, aconsejándose
realizar estudios prospectivos de biomarcadores.
Compuestos orgánicos volátiles
Los compuestos orgánicos volátiles (COV) son detectables
en el aliento exhalado y se han propuesto como
biomarcadores no invasivos para enfermedades como
el cáncer y, más recientemente, para la HAP94-96. Se han
identificado alteraciones significativas de los COV exhalados
entre pacientes con HAP, tanto en comparación
con controles como con pacientes con otras enfermedades
respiratorias, lo que sugiere que el análisis del
aliento exhalado tendría una posible aplicación médica
no invasiva en el campo de la HAP95.
Prueba de esfuerzo
Si bien los informes sobre la evolución natural de la
enfermedad vascular pulmonar temprana son limitados
y los pacientes suelen presentarse con enfermedad
avanzada97; sin embargo, se acepta generalmente que
el desarrollo de la HP inducida por el ejercicio precede
a la HP en reposo. Los estudios que utilizan ecocardiografía
de estrés han demostrado que los pacientes con riesgo de desarrollar HAP, como los familiares de
pacientes con HAPH e HAPI, tienen un aumento exagerado
de la PAPS en respuesta al esfuerzo98,99. Además,
los pacientes con HP por esfuerzo en el contexto de la
ES tienen un mayor riesgo de desarrollar HAP100. La
prueba de esfuerzo cardiopulmonar (PECP) también se
puede utilizar para identificar la causa de la disnea y
se implementa cada vez más en clínicas especializadas
en disnea. Los pacientes con HAP tienen anomalías
características que reflejan una limitación cardíaca
para el ejercicio asociada con una mayor ventilación
del espacio muerto e hiperventilación. Aunque la PECP
proporciona una gran cantidad de información fisiológica,
lleva mucho tiempo realizarla y requiere una gran
experiencia, lo que significa que su implementación se
limita a grupos de riesgo cuidadosamente seleccionados.
Un estudio de 895 pacientes con HP del Grupo 1
al Grupo 5 demostró que el 89% de los pacientes asintomáticos
(CF I según OMS), el 93% de los pacientes
con disnea leve (CF II según OMS) y el 100% de los
pacientes en CF III y IV según OMS caminaban <80%
de su capacidad de ejercicio predicha, utilizando una
PM6M; esta prueba sería más adecuada para la detección
de grandes poblaciones101.
Diagnóstico por imágenes
Siguen habiendo avances significativos en las tecnologías
de imágenes y también un retraso entre el informe
de la evidencia que demuestra el valor diagnóstico
de las imágenes y su implementación en la práctica
clínica3,47,102. En consecuencia, existe un beneficio de
diagnóstico potencial significativo que se puede realizar
para ayudar al diagnóstico más temprano de HAP. Muchas
características de la HP visibles en una tomografía
computarizada (TC) realizada para la evaluación de la
disnea inexplicable con frecuencia no son informadas
por los radiólogos ni reconocidas por los médicos.
Agrandamiento de la arteria pulmonar, índice elevado
de arteria pulmonar/aorta, dilatación del VD, índice
elevado de VD/ventrículo izquierdo y anomalías de la
perfusión pulmonar, como vidrio esmerilado centrolobulillar,
que se observan en la HAP103, potencialmente
podría evaluarse utilizando enfoques de inteligencia
artificial (IA). Se ha demostrado que los enfoques de
aprendizaje automático mejoran la automatización y
la cuantificación de algunos parámetros de imágenes,
particularmente en imágenes de resonancia magnética104.
Existe un interés creciente en la aplicación de
estos enfoques en técnicas de imagen más accesibles,
como la TC, donde la aplicación de enfoques basados
en patrones, como los que se usan en el software de
reconocimiento facial, podría usarse para reconocer
modelos consistentes con un diagnóstico de HP.
Dado que la mayoría de los pacientes con HAP se
presentan con disnea, el desarrollo de clínicas especializadas,
donde médicos experimentados realicen
investigaciones diagnósticas de manera sistemática y
oportuna, podría reducir el tiempo hasta el diagnóstico
y también permitir potencialmente la evaluación de las
herramientas de detección mencionadas anteriormente.
El uso de enfoques de IA para los datos recopilados de
forma rutinaria es un área de creciente interés. En muchos
países del mundo existen grandes y complejos conjuntos
de datos sobre la utilización de los recursos sanitarios.
Si se pudieran desarrollar algoritmos predictivos para
identificar a los pacientes con mayor riesgo de HAP, los
beneficios potenciales podrían incluir oportunidades para
evaluar el impacto económico de las pruebas de diagnóstico
y el tratamiento, sin el posible sesgo de tiempo
de espera que existe con los enfoques actuales. Además,
los datos de pacientes en una etapa más temprana de la
enfermedad pueden brindar nuevos conocimientos sobre
los mecanismos moleculares tempranos y resaltar nuevos
objetivos para el desarrollo de fármacos.
Conclusiones
Existe evidencia convincente de que la detección
de HAP en poblaciones de alto riesgo permitirá un
diagnóstico e intervención terapéutica precoz con una
oportunidad invalorablemente prometedora para mejorar
el pronóstico de estos pacientes. Sin embargo, los
métodos de detección que se utilizan habitualmente en
la práctica clínica tienen limitaciones y es posible que
se requiera una combinación de herramientas o parámetros
para mejorar la sensibilidad y la selectividad de
los programas que utilizados. A pesar de la evidencia
del éxito de los algoritmos de detección para facilitar
el diagnóstico temprano de HAP en ES, el progreso en
el diagnóstico temprano de HAP en otras formas de
HAP, como la HAPI, sigue siendo decepcionante. La
creación de algoritmos de detección para pacientes con
riesgo de HAP con ES ha aumentado la velocidad y la
especificidad del diagnóstico, mejorando potencialmente
la sobrevida, aunque los costos siguen siendo
importantes, pero insignificantes ante los valores de las
terapéuticas necesarias para el tratamiento de la HAP
en etapas avanzadas.
Es necesario el desarrollo y la validación de algoritmos
de detección de HAP de otras etiologías si queremos
realizar mejoras similares para una población de pacientes
más amplia. La detección de HAP en pacientes
asintomáticos en riesgo y el desarrollo de enfoques basados
en la detección en pacientes sintomáticos, donde
el diagnóstico rara vez se considera, son necesarios
para mejorar las tasas de detección y reducir el tiempo
hasta el diagnóstico.
En resumen, aunque se han logrado avances, todavía
existe una necesidad clara e insatisfecha de mejoras en
el diagnóstico, caracterización y manejo de los pacientes
con HAP. Un diagnóstico definitivo inicial a menudo
se retrasa hasta 2 años desde el inicio de los síntomas
y sigue siendo un desafío importante.
La HAP es una enfermedad que progresa rápidamente, incluso en pacientes con síntomas leves, y la intervención
terapéutica oportuna es esencial para influir en el
pronóstico a largo plazo de estos pacientes.
Recursos financieros
Los autores no recibieron ningún apoyo económico
para la investigación.
Conflicto de intereses
Los autores declararon no tener conflicto de intereses.
Referencias bibliográficas
1. Bevacqua RJ, Perrone SV. Manifestaciones pulmonares y
sistémicas de la hipertensión arterial pulmonar. Un enfoque
panvascular. Insuf Card 2021;16(1):14-36.
2. Galiè N, Humbert M, Vachiéry JL, Gibbs S, Lang I, Torbicki
A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M,
Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W,
Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade
PT, Zompatori M, Hoeper M; ESC Scientific Document Group.
2015 ESC/ERS Guidelines for the diagnosis and treatment of
pulmonary hypertension: the Joint Task Force for the Diagnosis
and Treatment of Pulmonary Hypertension of the European
Society of Cardiology (ESC) and the European Respiratory
Society (ERS): Endorsed by: association for European
Paediatric and Congenital Cardiology (AEPC), International
Society for Heart and Lung Transplantation (ISHLT). Eur Heart
J 2016;37:67-119.
3. Kiely DG, Lawrie A, Humbert M. Screening strategies for
pulmonary arterial hypertension. Eur Heart J Suppl 2019; 21
(Suppl K): K9-K20.
4. Beshay S, Sahay S, Humbert M. Evaluation and management of
pulmonary arterial hypertension. Respir Med 2020;171:106099.
5. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical
classification of pulmonary hypertension. J Am Coll Cardiol
2013;62(25 suppl):D34-D41.
6. Bourgeois A, Omura J, Habbout K, et al. Pulmonary arterial
hypertension: new pathophysiological insights and emerging
therapeutic targets. Int J Biochem Cell Biol 2018;104:9-13.
7. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic
definitions and updated clinical classification of pulmonary
hypertension. Eur Respir J 2019;53(1):1801913.
8. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial
hypertension in France: results from a national registry. Am J
Respir Crit Care Med 2006; 173: 1023-1030.
9. Humbert M, Coghlan JG, Khanna D. Early detection and
management of pulmonary arterial hypertension. Eur Respir
Rev 2012; 21: 126, 306-312.
10. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients
with idiopathic, familial, and anorexigen-associated pulmonary
arterial hypertension in the modern management era. Circulation
2010; 122: 156-163.
11. Thenappan T, Shah SJ, Rich S, et al. Survival in pulmonary
arterial hypertension: a reappraisal of the NIH risk stratification
equation. Eur Respir J 2010; 35: 1079-1087.
12. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors
for death in systemic sclerosis: a study from the EULAR
Scleroderma Trials and Research (EUSTAR) database. Ann
Rheum Dis 2010; 69:1809-1815.
13. Humbert M, Sitbon O, Yaïci A, et al. Survival in incident
and prevalent cohorts of patients with pulmonary arterial
hypertension. Eur Respir J 2010; 36: 549-555.
14. Condliffe R, Kiely DG, Peacock AJ, et al. Connective tissue
disease associated pulmonary arterial hypertension in the
modern treatment era. Am J Respir Crit Care Med 2009; 179:
151-157.
15. Dimopoulos K, Inuzuka R, Goletto S, et al. Improved survival
among patients with Eisenmenger syndrome receiving advanced
therapy for pulmonary arterial hypertension. Circulation 2010;
121:20-25.
16. Humbert M, Yaïci A, de Groote P, et al. Screening for pulmonary
arterial hypertension in patients with systemic sclerosis: clinical
characteristics at diagnosis and long-term survival. Arthritis
Rheum 2011; 63: 3522-3530.
17. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the
diagnosis and treatment of pulmonary hypertension. Eur Respir
J 2009; 34: 1219-1263.
18. Gibbs JSR. Making a diagnosis in PAH. Eur Respir Rev 2007;
16: 8-12.
19. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary
hypertension. A national prospective study. Ann Intern Med
1987; 107: 216-223.
20. Mazzei JA, Caneva JO, Perrone SV, Mellero MJ, Scali JJ,
Bortman G. Update in the diagnosis and treatment of pulmonary
hypertension. Medicina (Buenos Aires) 2011; 71(Supl. 1): 1-48.
21. Bevacqua RJ, Perrone SV. El impacto de CoViD-19 en la
hipertensión arterial pulmonar. Insuf Card 2021;16(3): 79-89
22. Lauga A, D’Ortencio A. Monitoreo de las presiones de la
arteria pulmonar: catéter de Swan-Ganz. Parte I. Insuf Card
2007;2(1):5-11.
23. Pérez-Olivares Delgado C, Aurtenetxe Pérez A, Escribano
Subías P. Hipertensión arterial pulmonar: progresión, pronóstico
e impacto. Insuf Card 2019; 14(Supl. 1): 1-7.
24. Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, Castillo-Palma MJ, Segovia
J, Gomez-Sanchez MA, Barbera JA; REHAP investigators.
Survival in pulmonary hypertension in Spain: insights from the
Spanish registry. Eur Respir J 2012;40:596-603.
25. Bevacqua RJ, Perrone SV. Avances futuros en la hipertensión
arterial pulmonar. Insuf Card 2013;8(4):185-190.
26. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension.
Circulation 2006;114:1417-1431.
27. Thierer J. Importancia del diagnóstico precoz en la hipertensión
pulmonar. Insuf Card 2009;4(2):52-58.
28. Galiè N, Torbicki A, Barst R, Dartevelle P, Haworth S,
Higenbottam T, Olschewski H, Peacock A, Pietra G, Rubin
LJ, Simonneau G. Guidelines on diagnosis and treatment of
pulmonary arterial hypertension: The Task Force on Diagnosis
and Treatment of Pulmonary Arterial Hypertension of the
European Society of Cardiology. Eur Heart J 2004;25:2243-2278.
29. Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ,
Olschewski H, Gaine S. Diagnosis and differential assessment of
pulmonary arterial hypertension. J Am Coll Cardiol 2004;43(12
Suppl S):40S-47S.
30. Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet
1998;352;9129:719-725.
31. D’Alonzo GE, Bower JS, Dantzker DR. Differentiation
of patients with primary and thromboembolic pulmonary
hypertension. Chest 1984;85;4:457-461.
32. Ahearn GS, Tapson VF, Rebeiz A, Greenfield JC, Jr.
Electrocardiography to define clinical status in primary
pulmonary hypertension and pulmonary arterial hypertension
secondary to collagen vascular disease. Chest 2002;122;2:524-527.
33. Bortman G. Presentación clínica y clasificación actual de la
hipertensión arterial pulmonar. Insuf Card 2009; 4(1):27-32.
34. Melero MJ. Estado actual de la hipertensión arterial pulmonar.
Insuf Card 2009; 4(1):23-26.
35. Raymond RJ, Hinderliter AL, Willis PW, Ralph D, Caldwell EJ,
Williams W, Ettinger NA, Hill NS, Summer WR, de Boisblanc
B, Schwartz T, Koch G, Clayton LM, Jobsis MM, Crow JW,
Long W. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol
2002;39;7:1214-1219.
36. Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita
M, Nakanishi N, Miyatake K. Clinical correlates and prognostic
significance of six-minute walk test in patients with primary
pulmonary hypertension. Comparison with cardiopulmonary
exercise testing. Am J Respir Crit Care Med 2000;161(2
Pt1):487-492.
37. Rubin LJ. Diagnosis and management of pulmonary arterial
hypertension: ACCP evidence-based clinical practice guidelines.
Chest 2004;126(1 Suppl):7S-10S.
38. Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary
function in primary pulmonary hypertension. J Am Coll Cardiol
2003;41;6:1028-1035.
39. Weir EK, Rubin LJ, Ayres SM, Bergofsky EH, Brundage
BH, Detre KM, Elliott CG, Fishman AP, Goldring RM,
Groves BM, et al. The acute administration of vasodilators
in primary pulmonary hypertension. Experience from the
National Institutes of Health Registry on Primary Pulmonary
Hypertension. Am Rev Respir Dis 1989;140;6:1623-1630.
40. Hachulla E, Gressin V, Guillevin L, et al. Early detection of
pulmonary arterial hypertension in systemic sclerosis: a French
nationwide prospective multicenter study. Arthritis Rheum
2005; 52: 3792-3800.
41. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA
2009 expert consensus document on pulmonary hypertension
a report of the American College of Cardiology Foundation
Task Force on Expert Consensus Documents and the American
Heart Association developed in collaboration with the American
College of Chest Physicians, American Thoracic Society, and
the Pulmonary Hypertension Association. J Am Coll Cardiol
2009; 53: 1573-1619.
42. Mukerjee D. Prevalence and outcome in systemic sclerosis
associated pulmonary arterial hypertension: application of a
registry approach. Ann Rheum Dis 2003; 62: 1088-1093.
43. Chung L, Domsic RT, Lingala B, et al. Survival and predictors
of mortality in systemic sclerosis-associated pulmonary arterial
hypertension: outcomes from the pulmonary hypertension
assessment and recognition of outcomes in scleroderma registry.
Arthritis Care Res 2014; 66: 489-495.
44. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry:
Assessing the Spectrum of Pulmonary hypertension Identified
at a REferral centre. Eur Respir J 2012; 39: 945-955.
45. Hickey PM, Lawrie A, Condliffe R. Circulating protein
biomarkers in systemic sclerosis related pulmonary arterial
hypertension: a review of published data. Front Med 2018; 5:
175.
46. Bergemann R, Allsopp J, Jenner H, et al. High levels of
healthcare utilization prior to diagnosis in idiopathic pulmonary
arterial hypertension support the feasibility of an early
diagnosis algorithm: the SPHInX project. Pulm Circ 2018; 8:
2045894018798613.
47. Kiely DG, Doyle O, Drage E, et al. Utilising artificial
intelligence to determine patients at risk of a rare disease:
idiopathic pulmonary arterial hypertension. Pulm Circ 2019;
9: 2045894019890549.
48. Coghlan JG, Denton CP, Grünig E, et al. Evidence-based
detection of pulmonary arterial hypertension in systemic
sclerosis: the DETECT study. Ann Rheum Dis 2014; 73: 1340-1349.
49. Hao Y, Thakkar V, Stevens W, et al. A comparison of the
predictive accuracy of three screening models for pulmonary
arterial hypertension in systemic sclerosis. Arthritis Res Ther
2015; 17: 7.
50. Meune C, Avouac J, Airo` P, et al. Prediction of pulmonary
hypertension related to systemic sclerosis by an index based
on simple clinical observations. Arthritis Rheum 2011; 63:
2790-2796.
51. Schreiber BE, Valerio CJ, Keir GJ, et al. Improving the
detection of pulmonary hypertension in systemic sclerosis using
pulmonary function tests. Arthritis Rheum 2011; 63: 3531-3539.
52. Hachulla E, de Groote P, Gressin V, et al. The three-year
incidence of pulmonary arterial hypertension associated with
systemic sclerosis in a multicenter nationwide longitudinal study
in France. Arthritis Rheum 2009; 60: 1831-1839.
53. Parent F, Bachir D, Inamo J, et al. A hemodynamic study of
pulmonary hypertension in sickle cell disease. N Engl J Med
2011;365: 44-53.
54. Park J, Park MS, Kwon JH, et al. Preoperative
2D-echocardiographic assessment of pulmonary arterial
pressure in subgroups of liver transplantation recipients.
Anesth Pain Med (Seoul) 2021;16(4):344-352. doi:10.17085/
apm.21028.
55. Allanore Y, Borderie D, Avouac J, et al. High N-terminal probrain
natriuretic peptide levels and low diffusing capacity for
carbon monoxide as independent predictors of the occurrence
of precapillary pulmonary arterial hypertension in patients with
systemic sclerosis. Arthritis Rheum 2008; 58: 284-291.
56. Khanna D, Gladue H, Channick R, Chung L, Distler O, Furst DE,
Hachulla E, Humbert M, Langleben D, Mathai SC, Saggar R,
Visovatti S, Altorok N, Townsend W, FitzGerald J, McLaughlin
VV, Scleroderma F; Scleroderma Foundation and Pulmonary
Hypertension Association. Recommendations for screening and
detection of connective tissue disease-associated pulmonary
arterial hypertension. Arthritis Rheum 2013;65:3194-3201.
57. Huang D, Cheng YY, Chan PH, Hai J, Yiu KH, Tse HF, Wong
KL, Fan K, Li YW, Ng WL, Yim CW, Wong CJ, Tam LS,
Wong PCH, Wong CY, Ho CH, Leung AMH, Mok CC, Lam
H, Lau CS, Cheung T, Ho C, Law SWY, Chan EW, Yin LX,
Yue WS, Mok TM, Evora MA, Siu CW. Rationale and design
of the screening of pulmonary hypertension in systemic lupus
erythematosus (SOPHIE) study. ERJ Open Res 2018;4: 00135-
2017.
58. Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC,
Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of
pulmonary arterial hypertension. Eur Respir J 2019;53:1801899.
59. Graf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W,
Hodgson J, Liu B, Salmon RM, Southwood M, Machado RD,
Martin JM, Treacy CM, Yates K, Daugherty LC, Shamardina
O, Whitehorn D, Holden S, Aldred M, Bogaard HJ, Church
C, Coghlan G, Condliffe R, Corris PA, Danesino C, Eyries
M, Gall H, Ghio S, Ghofrani HA, Gibbs JSR, Girerd B,
Houweling AC, Howard L, Humbert M, Kiely DG, Kovacs
G, MacKenzie Ross RV, Moledina S, Montani D, Newnham
M, Olschewski A, Olschewski H, Peacock AJ, Pepke-Zaba
J, Prokopenko I, Rhodes CJ, Scelsi L, Seeger W, Soubrier F,
Stein DF, Suntharalingam J, Swietlik EM, Toshner MR, van
Heel DA, Vonk Noordegraaf A, Waisfisz Q, Wharton J, Wort
SJ, Ouwehand WH, Soranzo N, Lawrie A, Upton PD, Wilkins
MR, Trembath RC, Morrell NW. Identification of rare sequence
variation underlying heritable pulmonary arterial hypertension.
Nat Commun 2018;9:1416.
60. Fontecha MB, Anadón M del R, Mazzei JA, Fundia AF. Avances
en la genética de la hipertensión arterial pulmonar. Insuf Card
2020;15(1): 10-18.
61. Mazzei JA. Mecanismos fisiopatológicos involucrados y
diagnóstico de la hipertensión arterial pulmonar. Insuf Card
2009; 4(1):3-10.
62. Yang H, Zeng Q, Ma Y, Liu B, Chen Q, Li W, Xiong C, Zhou
Z. Genetic analyses in a cohort of 191 pulmonary arterial
hypertension patients. Respir Res 2018;19:87.
63. Montani D, Girerd B, Jaïs X, Laveneziana P, Lau EMT,
Bouchachi A, Hascoët S, Günther S, Godinas L, Parent F,
Guignabert C, Beurnier A, Chemla D, Hervé P, Eyries M,
Soubrier F, Simonneau G, Sitbon O, Savale L, Humbert M.
Screening for pulmonary arterial hypertension in adults carrying
a BMPR2 mutation. Eur Respir J 2021;58(1): 2004229.
64. Blenda M, Hadinnapola C, Haimel M, Coghlan G, Corris PA,
Gibbs JS, Kiely DG, Lawrie A, Peacock AJ, Pepke-Zaba J,
Trembath RC, Wharton J, Wilkins MR, Wort SJ, Graf S, Morrell
NW. The UK National Cohort Study of idiopathic and heritable
pulmonary arterial hypertension. Am J Respir Crit Care Med
2016;193:A7364.
65. Hadengue A, Benhayoun MK, Lebrec D, Benhamou JP.
Pulmonary hypertension complicating portal hypertension:
prevalence and relation to splanchnic hemodynamics.
Gastroenterology 1991;100: 520-528.
66. Sitbon O, Lascoux-Combe C, Delfraissy JF, Yeni PG, Raffi F,
De Zuttere D, Gressin V, Clerson P, Sereni D, Simonneau G.
Prevalence of HIV-related pulmonary arterial hypertension in
the current antiretroviral therapy era. Am J Respir Crit Care
Med 2008;177:108-113.
67. Zagolin M, Llancaqueo M. Hipertensión pulmonar: importancia
de un diagnóstico precoz y tratamiento específico. Rev Med
Clin Condes 2015; 26(3) 344-356.
68. Sawada H, Mitani Y, Nakayama T, Fukushima H, Kogaki S,
Igarashi T, Ichida F, Ono Y, Nakanishi T, Doi S, Ishikawa
S, Matsushima M, Yamada O, Saji T. Detection of pediatric
pulmonary arterial hypertension by school electrocardiography
mass screening. Am J Respir Crit Care Med 2019;199:1397-
1406.
69. Strange G, Stewart S, Celermajer DS, Prior D, Scalia GM,
Marwick TH, Gabbay E, Ilton M, Joseph M, Codde J, Playford
D. Threshold of pulmonary hypertension associated with
increased mortality. J Am Coll Cardiol 2019;73:2660-2672.
70. D’Alto M, Romeo E, Argiento P, D’Andrea A, Vanderpool
R, Correra A, Bossone E, Sarubbi B, Calabro R, Russo MG,
Naeije R. Accuracy and precision of echocardiography versus
right heart catheterization for the assessment of pulmonary
hypertension. Int J Cardiol 2013;168: 4058-4062.
71. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A,
Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova
T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski
L, Spencer KT, Tsang W, Voigt JU. Recommendations for
cardiac chamber quantification by echocardiography in adults:
an update from the American Society of Echocardiography and
the European Association of Cardiovascular Imaging. Eur Heart
J Cardiovasc Imaging 2015;16:233-270.
72. Cursack G, Núñez C, Coronel ML, Echazarreta D, Lobo
Márquez LL, Lema L, Escudero E, Perna E. Disfunción grave de
ventrículo derecho por ecocardiografía en hipertensión arterial
pulmonar: prevalencia, predictores clínicos, ecocardiográficos
y tratamiento. Insuf Card 2017; 12(1): 16-23.
73. Tazar J, Haurigot MP, Caram R(h), Caram R, Haurigot GE.
Función sistólica del ventrículo derecho en pacientes con
hipertensión pulmonar Análisis con strain y strain rate. Insuf
Card 2012; 7(3):109-116.
74. Trip P, Nossent EJ, de Man FS, van den Berk IAH, Boonstra
A, Groepenhoff H, Leter EM, Westerhof N, Gru¨nberg K,
Bogaard H-J, Vonk-Noordegraaf A. Severely reduced diffusion
capacity in idiopathic pulmonary arterial hypertension:
patient characteristics and treatment responses. Eur Respir J
2013;42:1575-1585.
75. Giannakoulas G, Mouratoglou SA, Gatzoulis MA, Karvounis
H. Blood biomarkers and their potential role in pulmonary
arterial hypertension associated with congenital heart disease.
a systematic review. Int J Cardiol 2014;174:618-623.
76. Jardim C, Souza R. Biomarkers and prognostic indicators
in pulmonary arterial hypertension. Curr Hypertens Rep
2015;17:556.
77. Yildiz M, Sahin A, Behnes M, Akin I. An expanding role of
biomarkers in pulmonary arterial hypertension. Curr Pharm
Biotechnol 2017;18:491-494.
78. Blyth KG, Groenning BA, Mark PB, Martin TN, Foster JE,
Steedman T, Morton JJ, Dargie HJ, Peacock AJ. NT-proBNP
can be used to detect right ventricular systolic dysfunction in pulmonary hypertension. Eur Respir J 2007;29:737-744.
79. Fijalkowska A, Kurzyna M, Torbicki A, Szewczyk G, Florczyk
M, Pruszczyk P, Szturmowicz M. Serum N-terminal brain
natriuretic peptide as a prognostic parameter in patients with
pulmonary hypertension. Chest 2006;129:1313-1321.
80. Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW,
Foreman AJ, Frost AE, McGoon MD, Pasta DJ, Selej M, Burger
CD, Frantz RP. Predicting survival in patients with pulmonary
arterial hypertension: the REVEAL risk score calculator 2.0 and
comparison with ESC/ERS based risk assessment strategies.
Chest 2019;156:323-337.
81. Galiè N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC,
Klepetko W, McGoon MD, McLaughlin VV, Preston IR,
Rubin LJ, Sandoval J, Seeger W, Keogh A. Updated treatment
algorithm of pulmonary arterial hypertension. J Am Coll Cardiol
2013;62:D60-D72.
82. Zhang CY, Ma LL, Wang LX. Relationship between serum uric
acid levels and ventricular function in patients with idiopathic
pulmonary hypertension. Exp Clin Cardiol 2013;18:e37-e39.
83. Dimitroulas T, Giannakoulas G, Dimitroula H, Sfetsios T,
Parcharidou D, Karvounis H, Settas L. Significance of serum
uric acid in pulmonary hypertension due to systemic sclerosis:
a pilot study. Rheumatol Int 2011;31:263-267.
84. Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F,
Nakanishi N, Yamagishi M, Kunieda T, Miyatake K. Serum
uric acid levels correlate with the severity and the mortality of
primary pulmonary hypertension. Am J Respir Crit Care Med
1999;160:487-492.
85. San Miguel-Hernández A, Martín-Gil FJ, Armentia-Medina A.
Metodología y aplicaciones en proteómica clínica. Dial Traspl
2009;30(4):139-143.
86. McKee T, Mckee JR. Bioquímica. La base molecular de la vida.
Madrid: Mc Graw Hill, 2003; p. 661-90.
87. Zhou G, Chen T, Raj JU. MicroRNAs in pulmonary arterial
hypertension. Am J Respir Cell Mol Biol 2015;52:139-151.
88. Rothman AM, Chico TJ, Lawrie A. MicroRNA in pulmonary
vascular disease. Prog Mol Biol Transl Sci 2014;124:43-63.
89. Rhodes CJ, Wharton J, Boon RA, Roexe T, Tsang H, Wojciak-Stothard B, Chakrabarti A, Howard LS, Gibbs JS, Lawrie A,
Condliffe R, Elliot CA, Kiely DG, Huson L, Ghofrani HA,
Tiede H, Schermuly R, Zeiher AM, Dimmeler S, Wilkins MR.
Reduced microRNA-150 is associated with poor survival in
pulmonary arterial hypertension. Am J Respir Crit Care Med
2013;187:294-302.
90. Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan
L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW,
Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension.
J Clin Invest 2016;126:2495-2508.
91. Rhodes CJ, Wharton J, Ghataorhe P, Watson G, Girerd B,
Howard LS, Gibbs JSR, Condliffe R, Elliot CA, Kiely DG,
Simonneau G, Montani D, Sitbon O, Gall H, Schermuly
RT, Ghofrani HA, Lawrie A, Humbert M, Wilkins MR.
Plasma proteome analysis in patients with pulmonary arterial
hypertension: an observational cohort study. Lancet Respir Med
2017;5:717-726.
92. Rhodes CJ, Ghataorhe P, Wharton J, Rue-Albrecht KC,
Hadinnapola C, Watson G, Bleda M, Haimel M, Coghlan G,
Corris PA, Howard LS, Kiely DG, Peacock AJ, Pepke-Zaba J,
Toshner MR, Wort SJ, Gibbs JSR, Lawrie A, Gra¨f S, Morrell
NW, Wilkins MR. Plasma metabolomics implicates modified
transfer RNAs and altered bioenergetics in the outcomes of
pulmonary arterial hypertension. Circulation 2017;135:460-475.
93. Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz
RP, Foreman AJ, Badesch DB, McGoon MD. The REVEAL
Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012;141:354-362.
94. Cohen-Kaminsky S, Nakhleh M, Perros F, Montani D, Girerd
B, Garcia G, Simonneau G, Haick H, Humbert M. A proof of
concept for the detection and classification of pulmonary arterial
hypertension through breath analysis with a sensor array. Am J
Respir Crit Care Med 2013;188:756-759.
95. Nakhleh MK, Haick H, Humbert M, Cohen-Kaminsky S.
Volatolomics of breath as an emerging frontier in pulmonary
arterial hypertension. Eur Respir J 2017;49:1601897.
96. Nakhleh MK, Amal H, Jeries R, Broza YY, Aboud M, Gharra
A, Ivgi H, Khatib S, Badarneh S, Har-Shai L, Glass-Marmor
L, Lejbkowicz I, Miller A, Badarny S, Winer R, Finberg J,
Cohen-Kaminsky S, Perros F, Montani D, Girerd B, Garcia G,
Simonneau G, Nakhoul F, Baram S, Salim R, Hakim M, Gruber
M, Ronen O, Marshak T, Doweck I, Nativ O, Bahouth Z, Shi DY,
Zhang W, Hua QL, Pan YY, Tao L, Liu H, Karban A, Koifman
E, Rainis T, Skapars R, Sivins A, Ancans G, Liepniece-Karele
I, Kikuste I, Lasina I, Tolmanis I, Johnson D, Millstone SZ,
Fulton J, Wells JW, Wilf LH, Humbert M, Leja M, Peled N,
Haick H. Diagnosis and classification of 17 diseases from 1404
subjects via pattern analysis of exhaled molecules. ACS Nano
2017;11:112-125.
97. Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer
J, Benjamin N, Olsson KM, Meyer K, Vizza CD, Vonk-Noordegraaf A, Distler O, Opitz C, Gibbs JSR, Delcroix M,
Ghofrani HA, Huscher D, Pittrow D, Rosenkranz S, Grünig
E. Mortality in pulmonary arterial hypertension: prediction by
the 2015 European pulmonary hypertension guidelines risk
stratification model. Eur Respir J 2017;50:1700740.
98. Grunig E, Weissmann S, Ehlken N, Fijalkowska A, Fischer C,
Fourme T, Galiè N, Ghofrani A, Harrison RE, Huez S, Humbert
M, Janssen B, Kober J, Koehler R, Machado RD, Mereles
D, Naeije R, Olschewski H, Provencher S, Reichenberger F,
Retailleau K, Rocchi G, Simonneau G, Torbicki A, Trembath
R, Seeger W. Stress Doppler echocardiography in relatives
of patients with idiopathic and familial pulmonary arterial
hypertension: results of a multicenter European analysis of
pulmonary artery pressure response to exercise and hypoxia.
Circulation 2009;119:1747-1757.
99. Montani D, Dorfmuller P, Girerd B, Le Pavec J, Fadel E,
Simonneau G, Sitbon O, Humbert M. Natural history over
8 years of pulmonary vascular disease in a patient carrying
biallelic EIF2AK4 mutations. Am J Respir Crit Care Med
2018;198:537-541.
100. Dumitrescu D, Nagel C, Kovacs G, Bollmann T, Halank M,
Winkler J, Hellmich M, Grünig E, Olschewski H, Ewert R,
Rosenkranz S. Cardiopulmonary exercise testing for detecting
pulmonary arterial hypertension in systemic sclerosis. Heart
2017;103:774-782.
101. Billings CG, Lewis R, Armstrong IJ, Hurdman JA, Smith IA,
Austin M, Elliot CA, Charalampopoulos A, Sabroe I, Lawrie A,
Thompson AAR, Condliffe R, Kiely DG. Incremental shuttle
walking test distance is reduced in patients with pulmonary
hypertension in World Health Organization Functional Class
I. Front Med 2018;5:172.
102. Kiely D, Levin D, Hassoun P, Ivy DD, Jone PN, Bwika J, Kawut
SM, Lordan J, Lungu A, Mazurek J, Moledina S, Olschewski
H, Peacock A, Puri GD, Rahaghi F, Schafer M, Schiebler M,
Screaton N, Tawhai M, Van Beek EJ, Vonk-Noordegraaf A,
Vanderpool RR, Wort J, Zhao L, Wild J, Vogel-Claussen J,
Swift A. EXPRESS: statement on imaging and pulmonary
hypertension from the Pulmonary Vascular Research Institute
(PVRI). Pulm Circ 2019;doi:10.1177/2045894019841990.
103. Rajaram S, Swift AJ, Condliffe R, Johns C, Elliot CA, Hill
C, Davies C, Hurdman J, Sabroe I, Wild JM, Kiely DG. CT
features of pulmonary arterial hypertension and its major
subtypes: a systematic CT evaluation of 292 patients from the
ASPIRE Registry. Thorax 2015;70:382-387.
104. Pesapane F, Codari M, Sardanelli F. Artificial intelligence in
medical imaging: threat or opportunity? Radiologists again
at the forefront of innovation in medicine. Eur Radiol Exp
2018;2:35.